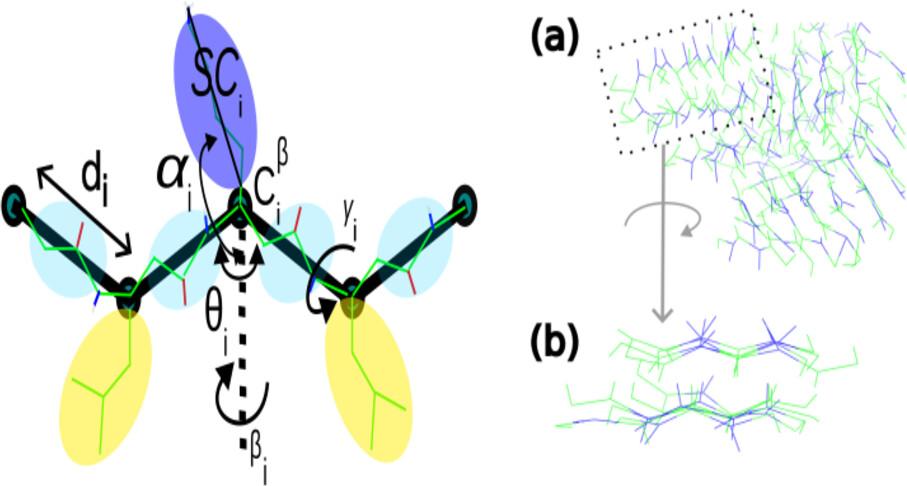
Coarse-grained simulations of foldamers such as β-peptides require force fields that accurately capture the backbone geometry and flexibility. In this work, we extend the UNRES coarse-grained model to β3-peptides by reparameterizing key local potential terms: virtual-bond stretching, virtual bond-angle bending, and torsional potentials. The bond-stretching term was derived from probability distributions obtained via all-atom molecular dynamics simulations of a reference β-peptide, while the angular and torsional potentials were fitted to quantum chemical potential energy surfaces computed by using the GFN2-xTB method with implicit solvent. Analytical potential forms were used to model the energy landscapes, and coefficients were obtained via nonlinear fitting to the potential of mean forces (PMFs). The modified UNRES model was validated through coarse-grained simulations and compared to the all-atom reference in terms of structural properties such as radius of gyration, end-to-end distances, and intramolecular side-chain separations. The capacity of the extended force field to reproduce β-peptide helical conformations was also evaluated with a peptide. Furthermore, the ability of the model to reproduce peptide self-assembly was evaluated using two peptides, one that is known to form large aggregates in aqueous solution and another that does not. The simulations successfully recapitulated these experimentally observed behaviors.